Annarita Miccio
Chromatin and gene regulation during development
Published on 22.10.2025
Presentation

Research areas
The laboratory of chromatin and gene regulation during development studies two main areas:
1. Dynamics of transcriptional and epigenetic networks during stem cell development
The definition of regulatory regions controlling the gene expression programs is fundamental for understanding the molecular mechanisms underlying many diseases and for the development of novel therapeutic approaches. Indeed, numerous disease-associated sequence variations occur in cis regulatory elements, which represent in some cases potential therapeutic targets. The aim of our projects is the genome-wide definition of the regulatory sequences used by human hematopoietic and epidermal stem/progenitor cells and their lineage-restricted progeny at different stages of development (Cavazza A., et al., Stem Cell Reports. 2016; Romano O. et al., Scientific Reports, 2016). The definition of the genetic and epigenetic programs was achieved through the use of a number of genomic and bioinformatic tools, including RNA-seq, deepCAGE, Retroviral scanning and ChIP-Seq. The outcome of this research is a better understanding of the molecular basis of stemness and lineage commitment of clinically relevant stem cells, which provides a knowledge basis for safer and more efficient usage of stem cells in cell and gene therapy (Romano O. et al., J Vis Exp. 2017; Romano O. et al., Stem Cells Translational Medicine, 2017).

Currently, we are analyzing genome-wide the occupancy of hematopoietic transcription factors (e.g., GATA1 and GATA2) and their co-factors, and the epigenetic histone modifications associated to transcription or silencing to define regulatory regions involved in hematopoietic stem cell biology and in erythroid commitment and differentiation. Validation of putative regulatory regions is performed by CRISPR-Cas9 targeted disruption and chromatic conformation capture assays (Romano et al., under revision; Meneghini et al., in preparation).

2. Molecular-based approaches for the treatment of β-Hemoglobinopathies
Sickle cell disease (SCD) and β-thalassemias are genetic diseases caused by mutations in the gene coding for the adult hemoglobin β-chain. They represent the most common monogenic disorders worldwide, affecting thousands of newborns annually. In β-thalassemia, the reduced production of adult β-chains causes α-globin precipitation and red blood cell death. In SCD, a single aminoacid substitution in the β-globin chain leads to polymerization of the sickle hemoglobin (HbS) and red blood cell deformation. β-globin disorders may lead to a severe clinical phenotype characterized by anemia, pain crises, and organ damage. So far, the only curative treatment is represented by bone marrow transplantation from a compatible donor, which, however, is available to less than 30% of the patients. Experimental treatments include gene therapy and pharmacological intervention. In the latter approach, efforts are underway to identify compounds that raise the expression of the fetal g-globin genes. The rational for this treatment is based on the long-standing observation that patients harboring mutations that trigger elevated g-globin expression, experience a more benign clinical course of the disease (Cavazzana, Antoniani and Miccio, Molecular Therapy, 2017). However, pharmacological treatments are not equally effective for all patients, are associated with a considerable toxicity and do not represent a definitive treatment. Several nuclear factors, such as the erythroid master regulator GATA1, its cofactors FOG1 and BCL11A and the NuRD repressor complex, are implicated in the silencing of g-globin expression. However, their role in erythroid development and hemoglobin switching has yet to be completely elucidated.

The goal of our research is to provide the basic scientific knowledge for developing safe therapies for SCD and β-thalassemias based on lentiviral and genome editing approaches aimed at increasing g-globin expression. We aim at characterizing the transcription factors and the regulatory genomic elements that control the switch from fetal to adult globin gene expression. The fine mapping of regulatory elements involved in hemoglobin switching can provide potential targets for therapeutic induction of fetal hemoglobin. Our studies are focused on the molecular mechanisms underlying the β-to-g-globin switching as well as on the evaluation of the efficacy and safety of these therapeutic approaches. We apply established and novel molecular techniques (e.g. genome-wide genomic analyses, lentiviral and CRISPR/Cas9 technologies) by using different cellular models, including clinically relevant hematopoietic stem cells (Antoniani, Meneghini et al., Blood, 2018; Lagresle-Peyrou C. et al., Haematologica. 2018; Lattanzi et al., Molecular Therapy, 2018; Weber et al., Molecular Therapy and Clinical Development, 2018; Weber, Frati et al., Science Advances, in press; Meneghini Ramadier et al., in preparation).
Projects
![]()
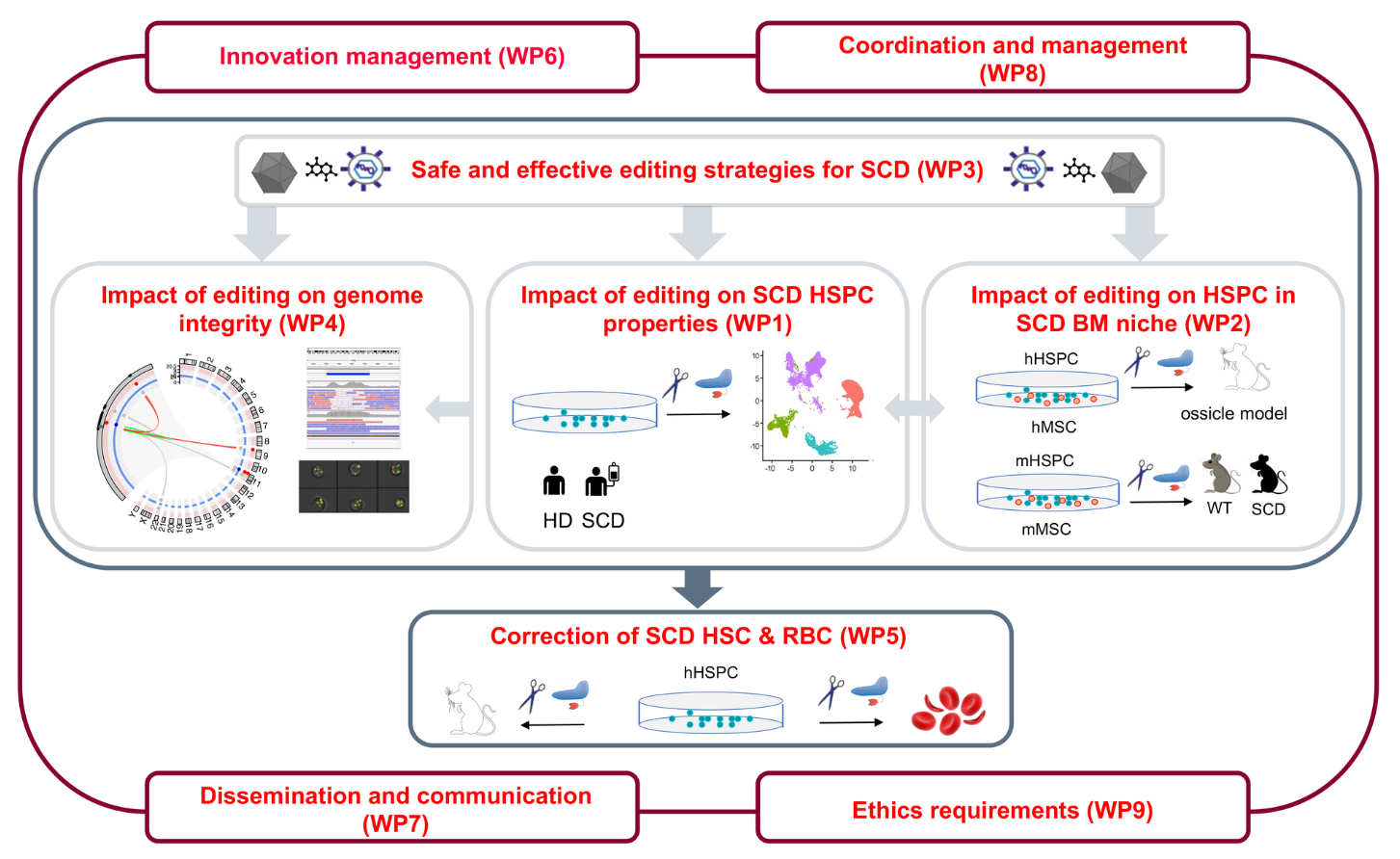
The EU-funded EDITSCD project aims to understand the molecular and cellular mechanisms underlying SCD HSPC dysfunctions and evaluate the impact of genome editing approaches on SCD HSPC. The objective of this study is to improve the SCD gene therapy strategy and evaluate the best tools and protocols for HSPC-based genome editing therapies.
Team













Resources & publications
-
 2025Journal (source)Blood
2025Journal (source)BloodA prime editing strategy to rewrite the γ-globin promoters and reactivate fet...
-
2025Journal (source)Nucleic Acids Research
Dissecting the epigenetic regulation of the fetal hemoglobin genes to unravel...
-
2023Journal (source)Mol Ther Nucleic Acids
Novel lentiviral vectors for gene therapy of sickle cell disease combining ge...
-
2023Journal (source)Blood
Adenine base editor-mediated correction of the common and severe IVS1-110 (G>...
-
2022Journal (source)Nat Commun
Base-editing-mediated dissection of a γ-globin cis-regulatory element for the...
-
2022Journal (source)Nat Med
Long-term outcomes of lentiviral gene therapy for the β-hemoglobinopathies: t...
-
2022Journal (source)Mol Ther
Combination of lentiviral and genome editing technologies for the treatment o...
-
2021Journal (source)Haematologica
Fetal hemoglobin rescues ineffective erythropoiesis in sickle cell disease.
-
2021Journal (source)Blood Adv
Correction of β-thalassemia by CRISPR/Cas9 editing of the α-globin locus in h...