Publié le 09.09.2025
L’une repose sur l’édition génétique des cellules souches sanguines, une approche prometteuse mais encore perfectible en termes de sécurité. En parallèle, l’équipe « Chromatine et régulation génique au cours du développement » dirigée par Annarita Miccio, développe une approche épigénétique, qui agit non pas sur le code de l’ADN, mais sur ses régulateurs : des marques chimiques qui contrôlent l’activation ou le silence des gènes. En modifiant précisément ces marques, on peut relancer l’expression de l’HbF sans toucher à l’ADN lui-même. Les travaux de Simone Amistadi et Letizia Fontana, publiés dans Nucleic Acid Research, montrent que cette méthode pourrait offrir une alternative sûre et efficace pour traiter ces maladies rares, tout en enrichissant notre compréhension des mécanismes qui régulent l’expression des gènes.
Les bêta-hémoglobinopathies sont des maladies génétiques graves, causées par des mutations du gène codant la chaîne de l'hémoglobine-β de l'adulte, la molécule de transport de l’oxygène. Les mutations entraînent la formation d’une hémoglobine malade, qui a pour conséquence une anémie et jusqu’aux lésions de plusieurs organes. La drépanocytose et la bêta-thalassémie, les deux bêta-hémoglobinopathies les plus fréquentes, sont considérées comme des troubles de la santé publique.
La gravité de la β-hémoglobinopathie peut être atténuée par une condition génétique bénigne appelée « persistance héréditaire de l'hémoglobine fœtale » (HPFH). Les mutations HPFH maintiennent la formation de l'hémoglobine fœtale (HbF) au-delà de la période embryonnaire.
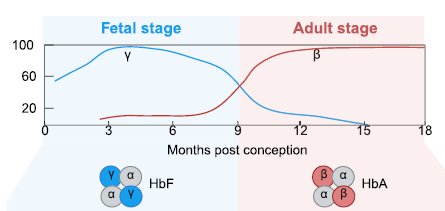
L’équipe d’Annarita Miccio, à l’Institut Imagine (Inserm, AP-HP, Université Paris Cité), travaille déjà sur des thérapies innovantes pour restaurer de manière contrôlée l’expression de l’hémoglobine fœtale chez les patients drépanocytaires. Leurs essais reposent actuellement sur l’édition du génome des cellules souches hémtopoïétiques, qui produisent les globules rouges. Néanmoins, en cas d’édition génétique, un certain nombre de complications sont à anticiper , comme les risques de mutations et de prolifération anormale des cellules hématopoïétiques une fois celles-ci modifiées.
C’est pourquoi, en parallèle des thérapies actuellement en développement, Simone Amistadi et Letizia Fontana ont également évalué la faisabilité de modifier l’expression de l’HbF par régulation épigénétique. L’épigénétique est l’ensemble des mécanismes qui régulent l’expression des gènes sans modifier la séquence de l’ADN. Ces mécanismes (par exemple, des marques chimiques dite « méthylation » sur l’ADN ou sur les protéines qui l’entourent) peuvent activer ou bloquer l’expression d’un gène sans altérer la séquence ADN.
L’édition épigénétique consiste à intervenir précisément sur ces marques régulatrices, pour moduler l’activité des gènes sans toucher au code génétique lui-même. Grâce à des outils dérivés des technologies comme CRISPR, on peut cibler un gène et, par exemple, "allumer" ou "éteindre" son expression de façon réversible. Cette approche ouvre des perspectives thérapeutiques prometteuses, notamment pour des maladies où certains gènes sont mal régulés sans être mutés. Elle est également plus sûre, car moins susceptible de causer des mutations secondaires lors de l’édition, qui pourraient entraîner des effets délétères à longue échéance.
La question de savoir si la méthylation de l'ADN influence directement l'expression de l'hémoglobine ou si cette extinction passe par des facteurs intermédiaires n'est toujours pas tranchée. C’est pourquoi les chercheurs ont caractérisé la régulation épigénétique du gène de la globine γ, en évaluant le rôle de la méthylation de l'ADN, et des modifications des histones (un type de petite protéine modifiant l’organisation du fil d’ADN) dans la modulation de son expression.
Leurs résultats, publiés récemment dans la prestigieuse revue scientifique Nucleic Acid Research, ont ainsi démontré que l'édition de l'épigénome représente une stratégie de pointe, efficace et sûre pour traiter les cellules souches hématopoïétiques des patients atteints de β-hémoglobinopathie (drépanocytose ou β-thalassémie) et pour obtenir une réactivation de l'HbF. Ces premiers travaux représentent ainsi une nouvelle piste de thérapie « génique » pour des maladies génétiques rares, et constituent également des travaux d’envergure dans la compréhension et la modulation de l’épigénome, un sujet encore vaste mais qui permettra de réduire l’errance diagnostique pour des patients encore en attente de l’identification de leur pathologie.
Référence :
Dissecting the epigenetic regulation of the fetal hemoglobin genes to unravel a novel therapeutic approach for β-hemoglobinopathies
S Amistadi, L Fontana et al., Nucleic Acid Research, 2025
Corresponding author : Annarita Miccio
https://doi.org/10.1093/nar/gkaf637