Published on 09.09.2025
Beta hemoglobinopathies are serious genetic diseases caused by mutations in the gene encoding the beta chain of adult hemoglobin, the oxygen-carrying molecule. The mutations lead to the formation of a diseased hemoglobin, resulting in anemia and even damage to several organs. Sickle cell anemia and beta-thalassemia, the two most common beta hemoglobinopathies, are considered public health disorders.
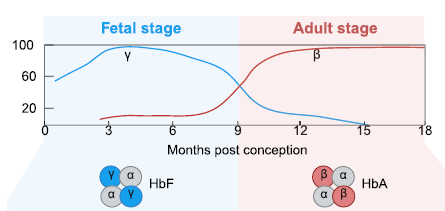
The severity of β-hemoglobinopathies can be mitigated by a benign genetic condition called “hereditary persistence of fetal hemoglobin” (HPFH). HPFH mutations result in the sustained expression of fetal hemoglobin (HbF) beyond the fetal period.
Annarita Miccio's team at Institut Imagine (Inserm, AP-HP, Université Paris Cité) is already working on innovative therapies to controllably restore fetal hemoglobin expression in sickle cell patients. Their trials are currently based on editing the genome of hematopoietic stem cells, which produce red blood cells. However, in the case of genetic editing, a number of complications need to be anticipated, such as the risk of mutations and abnormal proliferation of hematopoietic cells once they have been modified.
This is why, alongside the therapies currently under development, Simone Amistadi and Letizia Fontana have also assessed the feasibility of modifying HbF expression through epigenetic regulation. Epigenetics is the set of mechanisms that regulate gene expression without modifying the DNA sequence. These mechanisms (e.g. chemical marks called “methylation” on DNA or on the proteins surrounding it) can activate or block gene expression without altering the DNA sequence.
Epigenetic editing consists of intervening precisely on these regulatory marks to modulate gene activity without affecting the genetic code itself. Using tools derived from technologies such as CRISPR, we can target a gene and, for example, reversibly "switch on" or "switch off" its expression. This approach opens up promising therapeutic prospects, particularly for diseases where certain genes are poorly regulated without being mutated. It is also safer, as it is less likely to cause secondary mutations during editing, which could have deleterious long-term effects.
The question of whether DNA methylation directly influences hemoglobin expression, or whether this silencing is mediated by intermediate factors, remains unresolved. The researchers therefore characterized the epigenetic regulation of the globin γ gene, assessing the role of DNA methylation and histone modifications (a type of small proteins that modify the organization of the DNA strand) in modulating its expression.
Their results, recently published in the prestigious scientific journal Nucleic Acid Research, have demonstrated that epigenome editing represents a cutting-edge, effective and safe strategy for treating hematopoietic stem cells from patients with β-hemoglobinopathies (sickle cell disease or β-thalassemia) and for achieving HbF reactivation. This initial work represents a new avenue of “gene” therapy for rare genetic diseases and is also a major step forward in the understanding and modulation of the epigenome - still a vast subject, but one that will help to reduce diagnostic delays for patients still waiting for their pathology to be identified.
Référence:
Dissecting the epigenetic regulation of the fetal hemoglobin genes to unravel a novel therapeutic approach for β-hemoglobinopathies
S Amistadi, L Fontana et al., Nucleic Acid Research, 2025
Corresponding author: Annarita Miccio
https://doi.org/10.1093/nar/gkaf637