Publié le 08.04.2022
Aujourd’hui, seuls 50% des patients atteints de maladies génétiques ont un diagnostic. Une des ambitions de l’Institut Imagine est de hisser ce chiffre à 80% dans les prochaines années et réduire ainsi l’errance diagnostic des patients et leurs familles. Cela passe notamment par l’identification de nouvelles mutations génétiques. Or, dans une nouvelle publication parue le 6 avril 2022 dans la revue American Journal of Human Genetics, le laboratoire « Génétique des troubles du neurodéveloppement », dirigé par Vincent Cantagrel, a justement découvert un nouveau gène responsable d’une maladie rare du cervelet [1].
Cette étude a été initiée suite au suivi d’un premier patient (décédé à l’âge de 25 mois) présentant un tonus musculaire fortement réduit et un défaut du système nerveux autonome avec des troubles de la respiration et de la déglutition. Des examens IRM ont révélé une diminution de la taille de son cervelet, structure cérébrale sous corticale située à l’arrière du cerveau, essentielle dans des fonctions motrices comme l’apprentissage de la marche et l’équilibre, mais aussi dans des processus cognitifs de plus haut niveau tels que l’attention ou le langage. L’anomalie de développement touchait aussi le tronc cérébral, la partie du cerveau qui fait la jonction avec la moelle épinière.
Aucun diagnostic n’avait alors pu être établi. « En s’appuyant sur un panel de gènes déjà connus, les médecins n’ont pas pu identifier de mutations génétiques pouvant expliquer cette pathologie », résume Marion Coolen, investigatrice principale de la publication. Cela a poussé les chercheurs à réaliser un séquençage de l’exome, un examen consistant à passer en revue toutes les parties codantes de l’ADN du patient. « Nous avons ainsi repéré une même mutation dans les deux copies du gène PRDM13 », explique la chercheuse. Grâce à une collaboration avec le Centre de référence des malformations et maladies congénitales du cervelet, le CHU de Bordeaux, ainsi qu’avec des laboratoires étrangers, de nouveaux patients porteurs de mutations dans le gène PRDM13 ont été identifiés par la suite, avec des manifestations cliniques similaires.
Un rôle clé dans l'équilibre de la composition neuronale
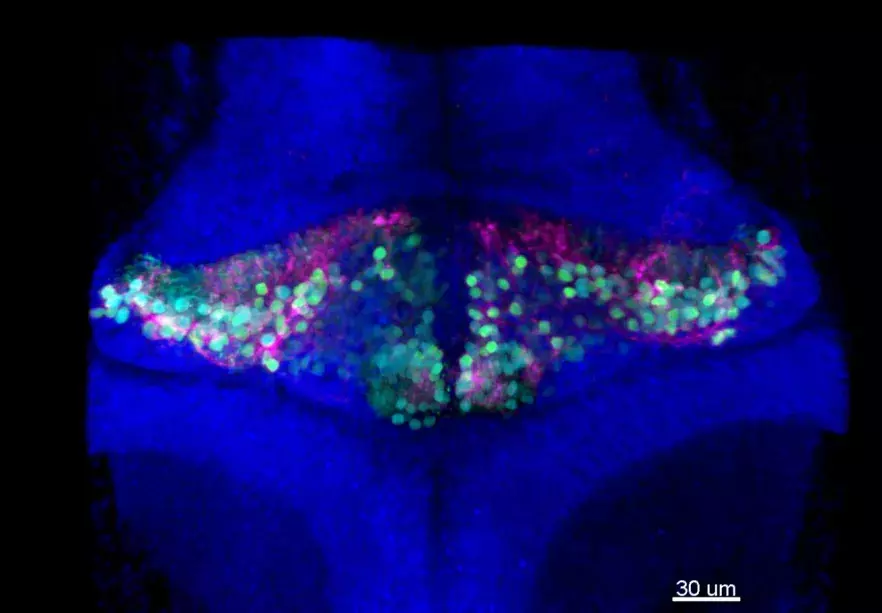
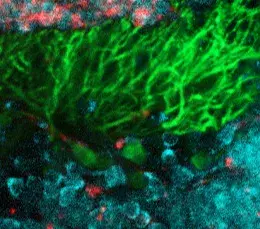
Bien que suspecte, cette corrélation ne permettait toutefois pas d’affirmer l’implication directe du gène dans la maladie. En effet aucun rôle n’était connu pour PRDM13 dans le développement embryonnaire du cervelet et du tronc cérébral. En combinant deux techniques (l’hybridation in situ et l’analyse transcriptomique en cellule unique - ou Single Cell), les chercheurs ont ensuite pu montrer que le PRDM13 s’exprime uniquement à des stades très précoces du développement, dans une région spécifique du cervelet où se forment les neurones dits « inhibiteurs », en particulier les cellules de Purkinje, très reconnaissables à leurs ramifications foisonnantes (voir photo). Le gène PRDM13 semble ainsi jouer un rôle central dans la différenciation des cellules immatures du cervelet en neurones inhibiteurs, et donc dans l’équilibre de la composition neuronale du cervelet.
Notre découverte a déjà eu un intérêt immédiat pour les familles
Afin de préciser les mécanismes physiopathologiques à l’œuvre, les chercheurs ont utilisé un autre modèle : le poisson-zèbre. « Son cervelet ressemble beaucoup au nôtre aussi bien dans sa structure que dans sa composition cellulaire. Par ailleurs, grâce aux analyses de données Single Cell réalisées en collaboration avec la plateforme de Bioinformatique et le laboratoire d’Antonio Rausell [de bio-informatique clinique], nous avons pu mettre en évidence de grandes similitudes dans l’expression de PRDM13 chez l’homme et le poisson zèbre au cours du développement. Ce qui en fait un bon modèle », explique Marion Coolen.
Le poisson zèbre pour mieux comprendre la maladie
Les chercheurs ont ensuite étudié une lignée de poissons présentant les mêmes mutations que le patient et ont pu observer une réduction importante du nombre de cellules de Purkinje dans le cervelet, exactement comme observé chez le patient. Les larves de poisson présentaient également des anomalies similaires du tronc cérébral. Autant d’arguments permettant de dire que la mutation de PRDM13 est bien responsable de la maladie.
« Notre découverte a déjà eu un intérêt immédiat pour les familles, indique la chercheuse. Ainsi une famille a déjà pu bénéficier d’un diagnostic prénatal à Necker qui a accompagné l’arrivée d’un enfant en bonne santé et d’autres familles sont demandeuses.»
Ce travail a aussi permis de mettre en lumière le rôle essentiel de PRDM13 dans le développement – encore méconnu – des structures impliquées dans la régulation des fonctions du cervelet et du système nerveux autonome. Ces structures pourraient être impliquées dans d’autres pathologies telle que la mort subite du nourrisson. Enfin ce travail pourrait aussi apporter un éclairage nouveau sur les programmes génétiques qui sous-tendent la spécification des types neuronaux et comment ces programmes se réadaptent en présence d’une mutation telle que celle des patients PRDM13.
Le laboratoire travaille actuellement à la mise au point d’un modèle de type organoïde, sorte de cervelet miniature reproduit in vitro à partir de cellules souches de patients, qui serait idéal pour approfondir la compréhension de ces mécanismes. Cela pourrait permettre d’identifier de nouveaux gènes et des séquences régulatrices clefs, potentiellement mutées chez des patients. Sur le long-terme, une meilleure connaissance de la neurogénèse dans le cervelet pourra être exploitée à des fins thérapeutiques.